QMS for Medical Device Manufacturers
The only eQMS that unifies QMSR quality processes and 21 CFR Part 11 training compliance in one integrated system — built for medical device manufacturers.


QMSR-Compliant Quality Management for Medical Device Manufacturers
eLeaP is the only Quality Management System with enterprise Learning Management built in — purpose-built for medical device manufacturers operating under the FDA’s Quality Management System Regulation (QMSR, 21 CFR Part 820, effective February 2, 2026) and ISO 13485:2016. Where other platforms require a separate LMS integrated via API, eLeaP automates personnel qualification across every quality process in a single pre-validated system. → Get a Demo.
How It Works:
When design outputs are approved or SOPs are revised, they automatically become training assignments with completion tracking. Design changes cascade required training to affected engineering and manufacturing roles. ISO 14971 risk control measures trigger training when updated. CAPAs and CAPA effectiveness checks drive targeted corrective training with documented verification — all fully traceable for FDA inspections and ISO 13485 audits.Our pre-validated platform deploys in weeks, with built-in 21 CFR Part 11 compliance and templates aligned to QMSR, ISO 13485:2016, and ISO 14971. Trusted by medical device manufacturers, combination product developers, and life sciences organizations operating under FDA oversight and international regulatory frameworks.
21 CFR Part 11 Document Control for Medical Device Operations
SOPs, work instructions, device specifications, test protocols, and Device Design File (DDF) documentation — all managed in one Part 11-compliant system with version-controlled approval workflows and automatic training deployment on release.
Document Control That Supports FDA Inspection Readiness
QMSR requires manufacturers to establish and maintain procedures for controlling all documents required by the quality system. eLeaP controls the full document lifecycle — from draft through approved through superseded — with 21 CFR Part 11-compliant electronic signatures, complete audit trails, and version control that prevents use of superseded documents in production.
What makes eLeaP different: when a document is approved and released, training automatically deploys to every affected role. Read-and-understood acknowledgments, comprehension assessments, and role-based training matrices maintain themselves without manual coordination. When an FDA inspector requests documentation records during an inspection, everything is in one system, versioned, and traceable — including the training history associated with each document revision. Learn more. Get a Demo.
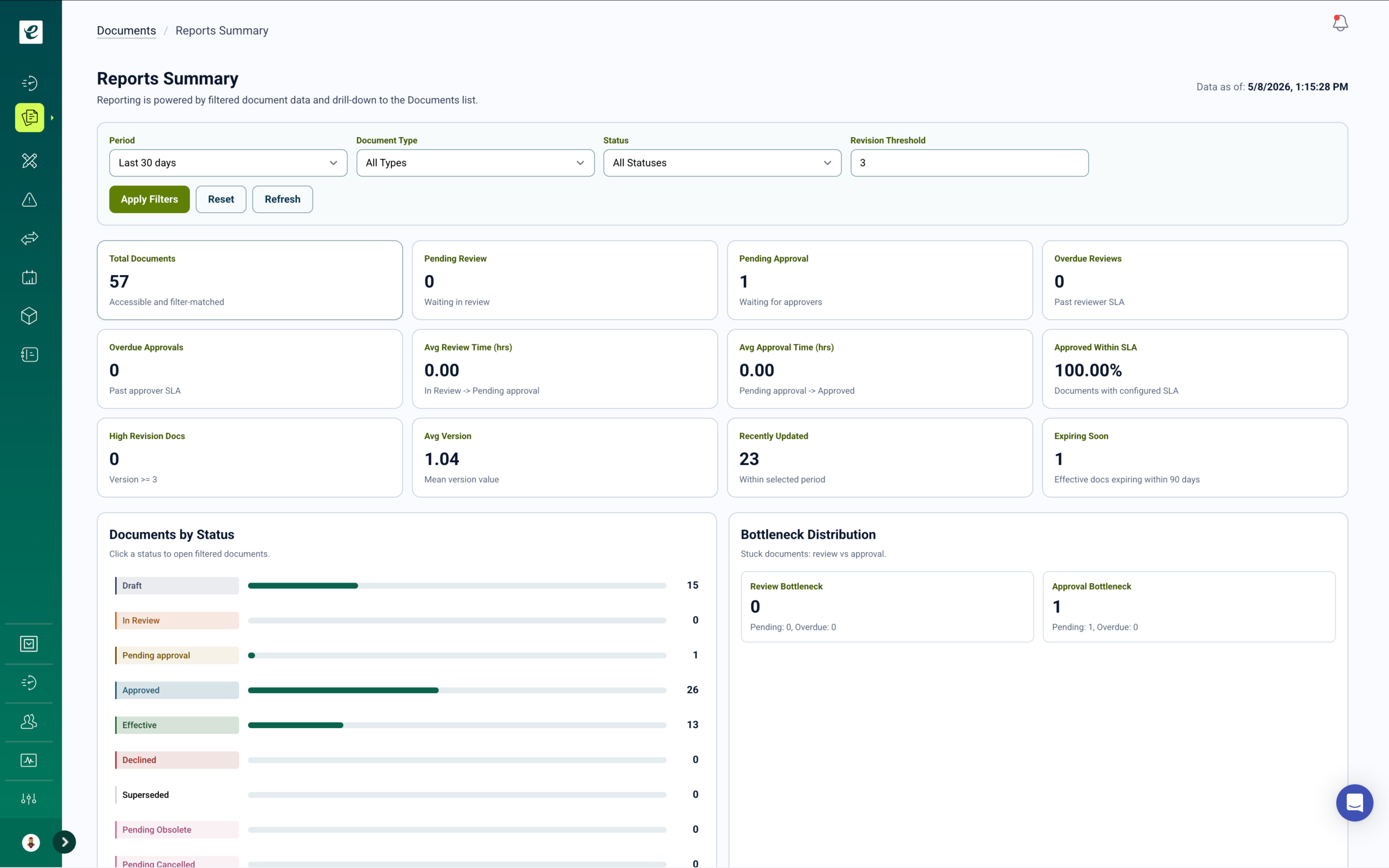
QMSR Design Controls with Integrated Competency Management
User needs, design inputs, design outputs, design verification, design validation, and design transfer — with full traceability from requirements through Device Design File (DDF) and Manufacturing Definition File (MDF) and automatic training as design phases progress.
Design Controls Built for QMSR and ISO 13485:2016 Compliance
QMSR aligns U.S. design control requirements with ISO 13485:2016 clause 7.3, establishing a structured design and development process with documented planning, inputs, outputs, reviews, verification, and validation. eLeaP’s design controls module manages requirements from initial user needs through design transfer and production release with complete traceability to support DDF and MDF compilation for regulatory submissions.
As design phases progress, training requirements automatically adjust. New design outputs trigger training assignments for affected engineering, manufacturing, and quality personnel. Design changes cascade training to impacted teams before implementation. Design reviews create verification records linked to training completion. Generate DDF and MDF documentation packages with complete training records, design review evidence, and approval documentation for regulatory submissions and technical files. Learn more.
ISO 14971 Risk Management with Integrated Training
Risk analysis, risk evaluation, risk control, and residual risk assessment for medical devices — aligned to ISO 14971:2019 — with automatic training deployment when risk control measures are introduced or updated.
When risk control measures are added or modified, training automatically deploys to affected design, manufacturing, and quality personnel. Root cause analysis connects quality events to training history, identifying whether issues stem from knowledge gaps, process variability, or device design failures. Risk reviews, control effectiveness monitoring, and CAPA linkage are maintained in one traceable system ready for FDA inspections and technical file submissions.
Risk Management That Connects to Every Quality Process
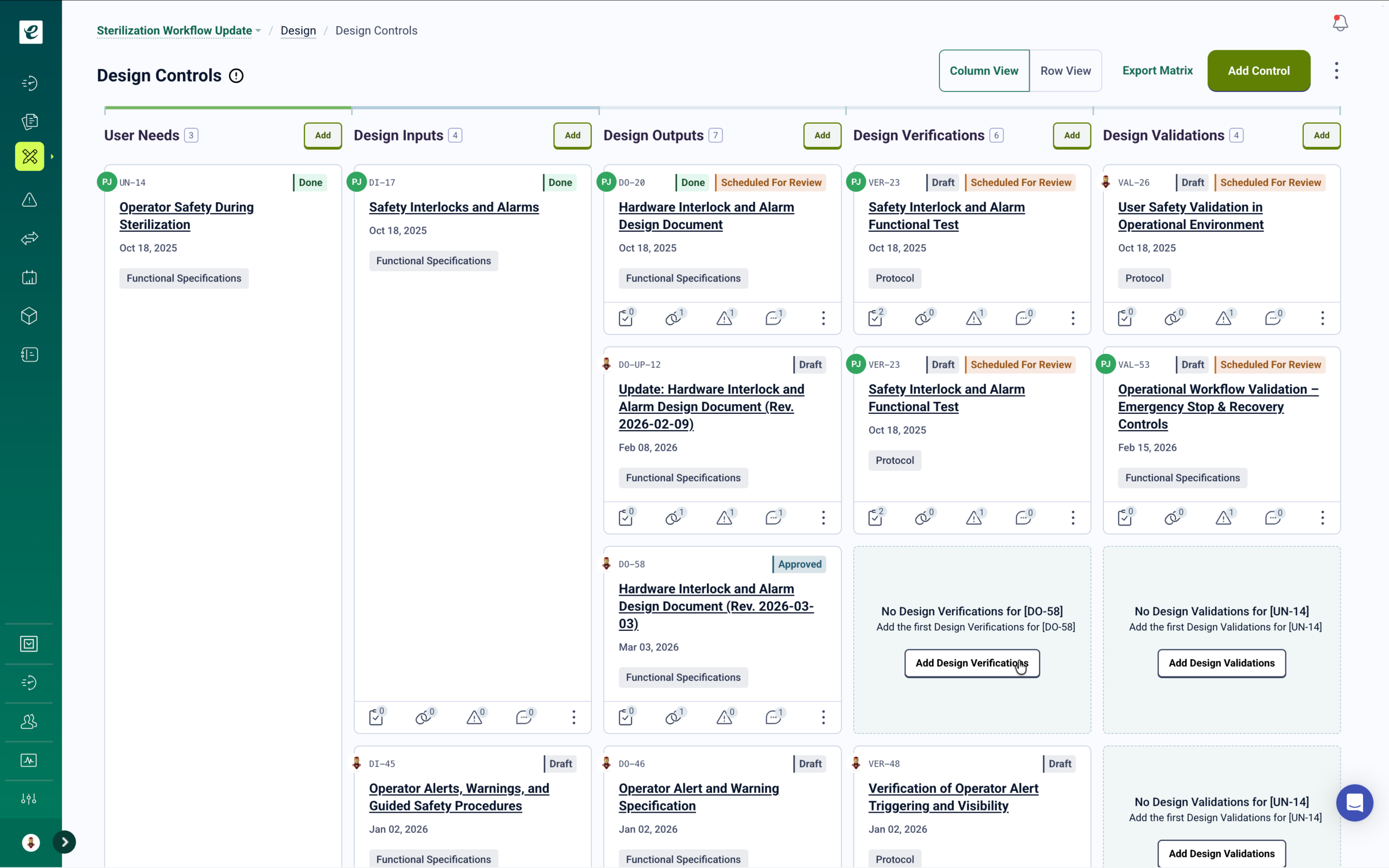
ISO 14971:2019 requires manufacturers to establish a risk management process throughout the device lifecycle. eLeaP’s risk management module supports hazard identification, risk estimation, risk evaluation, and risk control documentation with probability and severity scoring. Path-to-Harm visualization provides complete traceability from hazard through control measure through residual risk acceptance. Learn more.

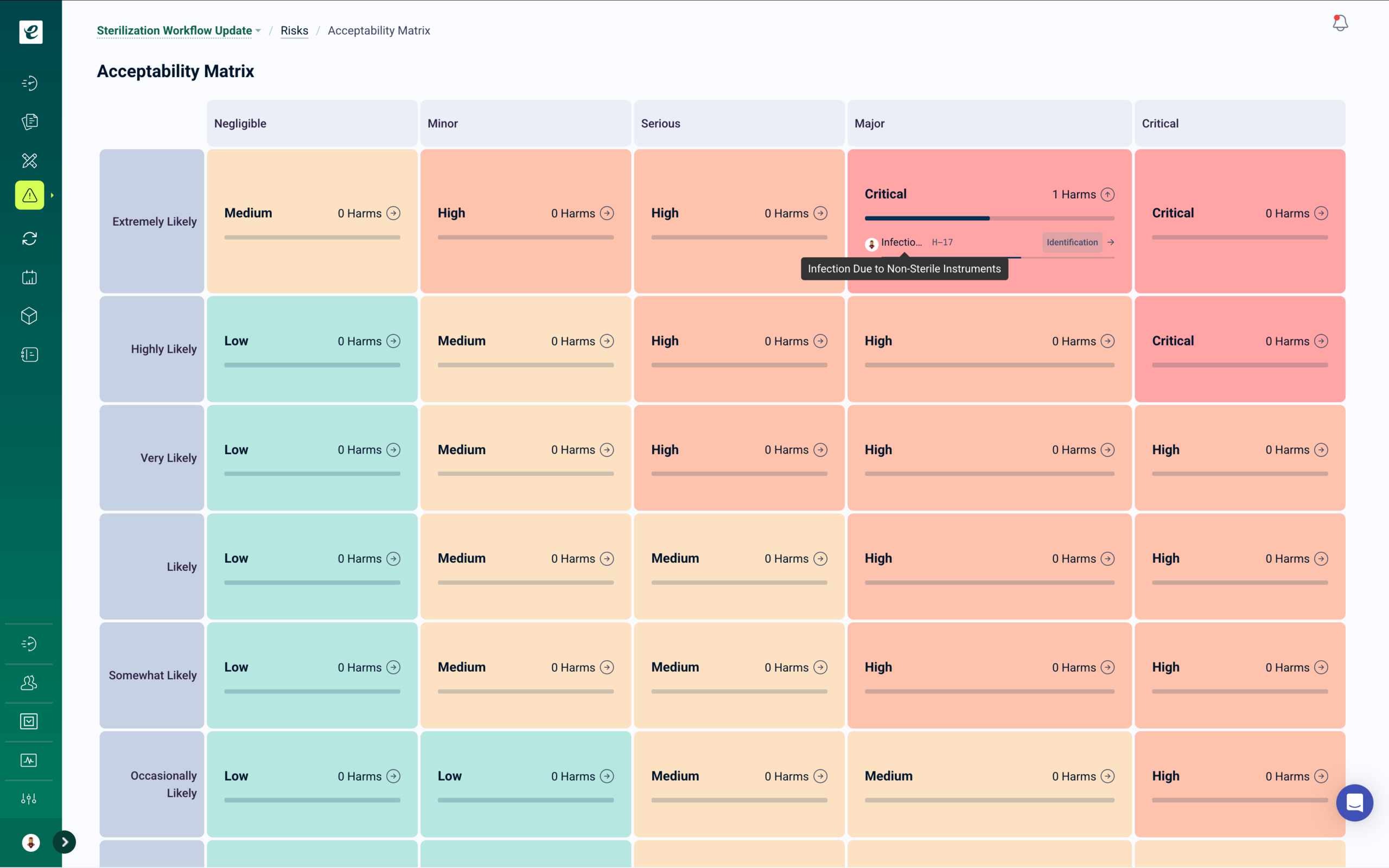
QMSR Change Control with Automatic Training
Design changes, manufacturing process modifications, software updates, and procedure revisions automatically trigger targeted training for affected personnel — with escalation workflows and documented implementation verification before changes are activated.
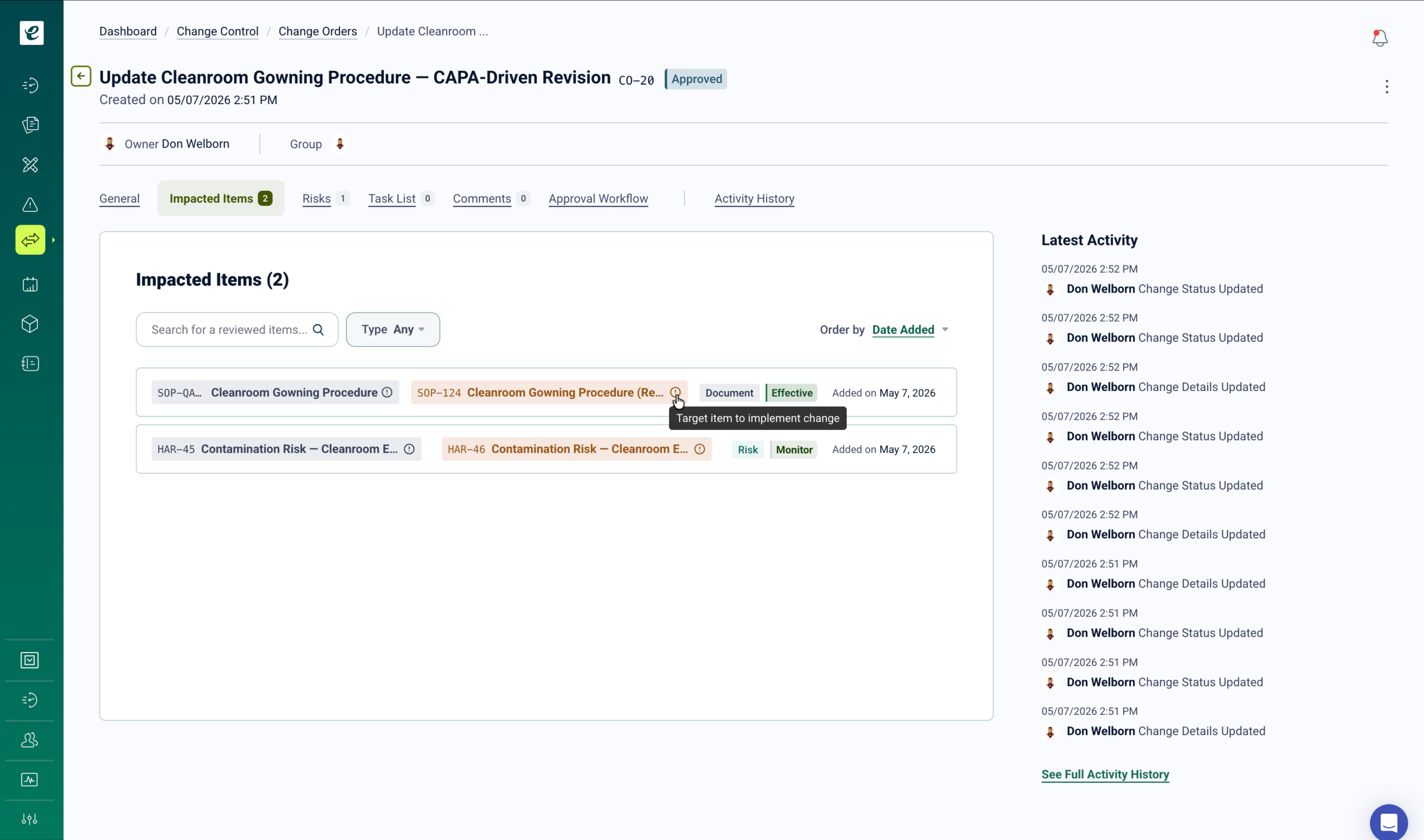
When a change is approved, training deploys immediately to all roles identified in the impact assessment. For design changes requiring FDA notification or submission, traceability from change request through training completion and implementation verification provides the regulatory evidence required. Change closure requires documented training completion — eliminating the compliance gap between change approval and verified personnel qualification. Complete change and training records populate directly into DDF and MDF documentation.
Change Control That Closes the Training Gap
QMSR requires manufacturers to establish and maintain procedures for change control, including documentation, review, and approval of all changes before implementation. Changes to device design may require submission to FDA and updated technical file documentation. eLeaP’s change control module manages the full change lifecycle from request through implementation and verification, with automated training deployment built into the approval workflow. Learn more.
CAPA, NCR, and MDR Event Management
Quality events — including CAPAs, nonconformances, complaint investigations, and Medical Device Reports — automatically identify training needs and deploy assignments, connecting investigations, corrective actions, and personnel competency in one traceable system.
From Quality Events to Verified Competency and Regulatory Compliance
QMSR requires documented processes for controlling nonconforming product, investigating quality events, implementing corrective and preventive actions, and reporting MDRs to FDA under 21 CFR Part 803. eLeaP’s events management module handles CAPAs, NCRs, complaint investigations, MDR determinations, and audit findings with structured workflows and automatic training deployment when root cause analysis identifies a personnel competency gap.
When a CAPA identifies a procedural misunderstanding or a complaint investigation reveals that operator training was inadequate, training assignments deploy automatically. CAPA closure requires documented training completion and effectiveness verification. MDR investigation records, complaint files, and corrective action evidence are maintained in one traceable system. All records are immediately available for FDA inspection and linked to the device’s quality history record. Learn more.
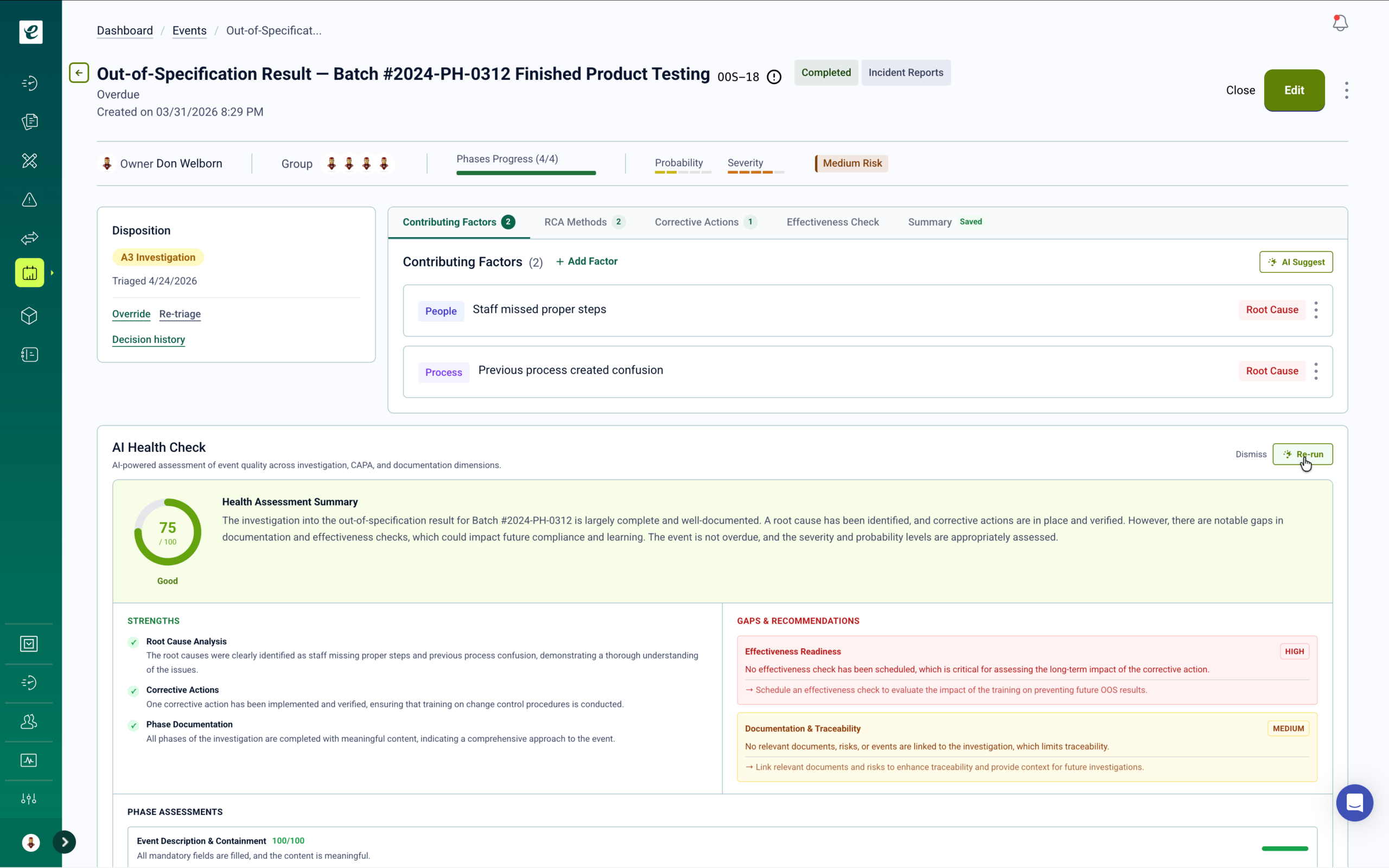
Supplier Controls for Medical Device Quality Systems
Approved supplier list management, supplier qualification, quality agreement documentation, and audit management — with training triggers when supplier audits identify gaps in internal qualification or receiving processes.
When supplier audits identify gaps in internal receiving, incoming inspection, or supplier qualification procedures, training automatically deploys to affected internal teams. Supplier performance metrics, qualification status, and audit findings are tracked in a system directly linked to your broader quality operations — and available for FDA inspection as part of your quality system records.
QMSR and ISO 13485 Supplier Control Requirements
QMSR and ISO 13485:2016 clause 7.4 require manufacturers to evaluate and select suppliers based on their ability to meet specified requirements, to maintain records of evaluation and re-evaluation results, and to establish appropriate controls over externally provided processes, products, and services. eLeaP’s supplier management module supports approved supplier list management, quality agreement documentation, incoming inspection procedures, supplier audit scheduling, and corrective action tracking. Learn more.
QMSR Training Compliance — Built In, Not Added On
Enterprise learning management system is native to the platform. QMSR training obligations, role-specific competency verification, and training effectiveness documentation are managed automatically — not tracked in a separate system or validated separately.
QMSR requires manufacturers to establish procedures for identifying training needs and ensuring that all personnel performing quality-affecting activities are trained. Training must be documented, competency must be verified, and records must be maintained. For FDA inspections, the relationship between training records and quality events — particularly CAPAs and design changes — is frequently reviewed. Meeting these requirements across engineering, manufacturing, QA, and regulatory personnel requires more than a training system that integrates with your QMS.
eLeaP’s learning management is native to the platform. Documents approved in Document Management automatically create training assignments. Design changes cascade training to impacted roles before implementation. CAPAs closed in Events automatically trigger effectiveness verification. Change Control approvals deploy role-based training before activation. Qualification matrices show who is trained on what — in real time, across every product line, procedure, and process. When an FDA inspector requests training records and asks to see the connection between a CAPA and subsequent training, you produce the complete record immediately — from one system.
Building QMSR Compliance Into Every Workflow
FDA inspections under QMSR consistently review training records, design control documentation, CAPA investigations, and the traceability between quality events and corrective actions. eLeaP eliminates inspection risk by making training an automatic output of every quality process — not a manual step requiring coordination between systems.
-
Automatic Training Triggers
Design changes, document approvals, CAPA closures, and change implementations automatically deploy training to the right personnel at the right time.
-
Real-Time Qualification Verification
Personnel qualification status is visible across all products, procedures, and processes — no spreadsheets, no manual tracking, no gaps when inspectors ask.
-
Inspection-Ready by Design
Complete audit trails connect quality decisions to training records. QMSR compliance documentation maintains itself as a natural output of your integrated system.
Quality and Training Working as One System
Medical device quality management must satisfy FDA expectations, ISO 13485 audit requirements, and international regulatory frameworks simultaneously. eLeaP gives quality and regulatory teams the tools to manage all three without running separate systems. Learn more.
-
Continuous Training Integration
Every quality process — documents, design, risk, change, events, suppliers — automatically drives training when it matters.
-
Connected Quality Data
All seven modules work as one system. No disconnected records. No manual handoffs. Complete audit trails across the device lifecycle.
-
Competency-Driven. Root Cause Analysis
Investigations connect training history, competency assessments, and quality outcomes — identifying whether issues stem from knowledge gaps, design failures, or process deficiencies.
-
Here, When You Need Us
Our quality experts support QMSR implementation, ISO 13485 certification preparation, and ongoing compliance program development for medical device manufacturers.