What is eQMS Software? Complete Guide for 2026

What Is eQMS Software? Complete Guide for 2026
An electronic Quality Management System (eQMS) is the software infrastructure through which regulated organizations manage the full lifecycle of their quality processes — from document control and CAPA management to change control, risk management, audit preparation, and training compliance. For quality managers operating under FDA, ISO, or GxP requirements, the distinction between a paper-based QMS and a purpose-built eQMS is not primarily about convenience. It is about whether the quality system can produce the audit evidence the regulatory environment demands, at the speed and precision regulators expect.
This guide explains what eQMS software is, which core modules it encompasses, how it maps to the regulatory frameworks governing regulated industries in 2026, and what quality managers need to evaluate when selecting a platform. It also addresses the integration gap between quality and training management that remains the most common documentation failure mode in regulated environments.
What Is eQMS Software?
eQMS software is a digital platform that centralizes and automates quality management processes within a validated, audit-trailed environment. Where a paper-based or manual QMS relies on physical documents, sign-in sheets, email-based approval chains, and disconnected spreadsheets, an eQMS provides a single controlled environment in which quality records are created, approved, executed, linked, and preserved.
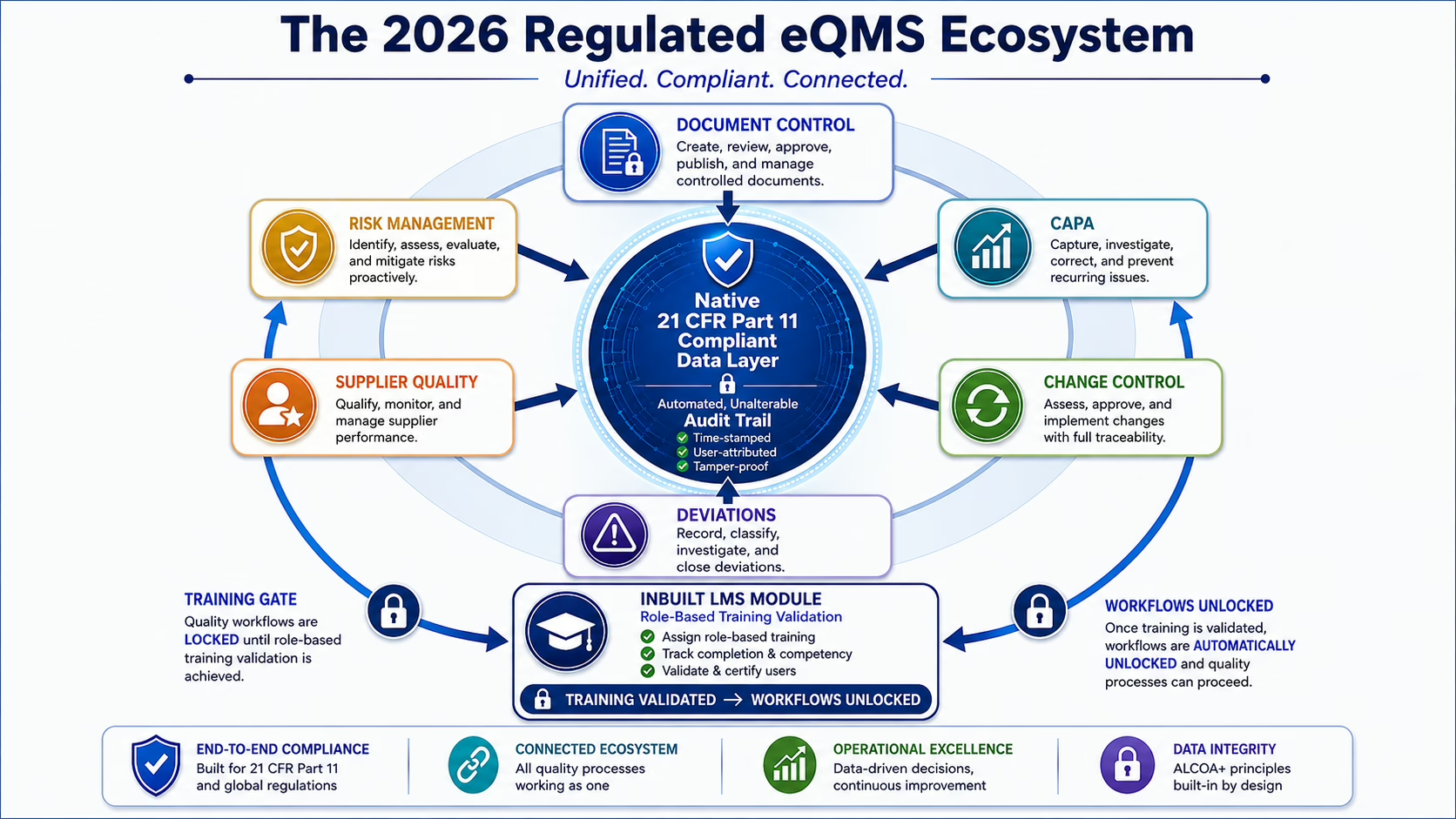
The defining characteristics of a regulated-industry eQMS are not the same as a general business process management tool. A regulated eQMS must satisfy specific technical requirements: electronic records that cannot be altered after approval without generating a visible audit trail, electronic signatures that are permanently and securely bound to the records they authenticate, controlled document versioning that prevents access to superseded versions after a new revision is approved, and record retention that meets the timeframes specified by applicable regulations.
21 CFR Part 11 — FDA’s regulation governing electronic records and electronic signatures in regulated industries — establishes the technical baseline that any eQMS deployed in a 21 CFR-regulated environment must satisfy. Part 11 requirements apply to electronic records that are created, modified, maintained, archived, retrieved, or transmitted under FDA requirements. An eQMS that does not satisfy Part 11 natively requires either manual workarounds or a separate validation effort to demonstrate compliance — both of which introduce risk and administrative overhead.
Core Modules of an eQMS Platform
Document Control
Document control is the foundation of any eQMS. It encompasses the creation, review, approval, distribution, and archiving of quality documents — standard operating procedures (SOPs), work instructions, quality policies, forms, and records — with version control that ensures personnel always work from the current approved version.
In a regulated environment, document control must satisfy specific requirements beyond basic file management. Under 21 CFR Part 11, approved documents must be protected against modification by unauthorized personnel, and any change to an approved record must generate an audit trail entry. Under QMSR (which amended 21 CFR Part 820 effective February 2, 2026)ISO 9001 and ISO 13485 Section 4.2.4, document control procedures must ensure documents are reviewed and approved prior to issue, that current versions are available at points of use, and that obsolete documents are prevented from unintended use.
An eQMS document control module automates the approval routing, controls access by role and document status, generates the Part 11-compliant audit trail, and creates the training trigger when a document revision is approved — linking document control directly to workforce competency management.
CAPA Management
Corrective and Preventive Action (CAPA) management is the quality process through which organizations identify the root cause of quality problems and implement systemic corrections that prevent recurrence. CAPA is an element of the Pharmaceutical Quality System under ICH Q10 Section 3.2.2, a requirement of QMSR under 21 CFR Part 820, and a core process under ISO 13485 Sections 8.5.2 and 8.5.3.
An eQMS CAPA module manages the full lifecycle: problem identification and description, root cause analysis with structured methodologies, corrective action planning with assigned owners and due dates, implementation verification, and effectiveness check at a defined interval after closure. Every step is timestamped and recorded in the audit trail, producing the documentation that regulators and auditors require to confirm that CAPAs are genuine systemic improvements rather than administrative closures.
The most operationally significant CAPA capability in an integrated eQMS is the automatic training trigger. When a CAPA identifies a training gap as a contributing root cause, or when the corrective action involves a procedure revision, the eQMS generates training assignments to affected personnel automatically. The CAPA record does not close until those training assignments are confirmed complete — creating a documented, enforced connection between the quality correction and the workforce competency response.
Change Control
Change control manages the review, approval, and implementation of changes to controlled processes, equipment, materials, facilities, and documentation. For pharmaceutical manufacturers, change control requirements derive from 21 CFR Parts 211.68 and 211.100 and from ICH Q10 Section 3.2.3. For medical device manufacturers, change control requirements are specified in QMSR and ISO 13485. For life sciences organizations broadly, changes to validated systems trigger additional obligations under 21 CFR Part 11 and applicable GxP predicate rules.
An eQMS change control module provides structured impact assessment workflows that route proposed changes to the appropriate reviewers based on the change category and scope. Approval chains are automated and documented. When a change results in a document revision, the system connects the change control record to the document update and to the training assignment generated by that revision — maintaining traceability from the change decision through document approval to confirmed workforce training.
Deviation and Nonconformance Management
Deviation and nonconformance management captures departures from approved procedures or specifications, documents the investigation and disposition, and tracks recurrence. In pharmaceutical manufacturing, the distinction between planned deviations (authorized in advance) and unplanned deviations (discovered after occurrence) carries regulatory significance and must be maintained in the quality record. Under ISO 13485 Section 8.3, nonconforming product must be identified and controlled to prevent unintended use or delivery.
An eQMS provides structured forms for deviation and nonconformance capture, investigation documentation, disposition records, and recurrence analysis. Links to CAPA records are maintained when a deviation generates a corrective action, creating an auditable chain from the initial event through to the systemic response.
Audit Management
Audit management in an eQMS covers the planning, execution, findings documentation, and closure of internal quality audits, supplier audits, and GMP compliance audits. ISO 9001:2015 Section 9.2 and ISO 13485 Section 8.2.4 both require internal audit programs with documented results. QMSR requires quality system audits under 21 CFR Part 820.
An eQMS audit management module provides scheduling tools, structured finding templates, classification and scoring of observations, and direct linkage from audit findings to CAPA records. When an internal audit identifies a training gap or a procedural nonconformance, the eQMS routes the finding into the corrective action workflow automatically. Follow-up audits reference the original findings and their disposition, maintaining continuity across audit cycles.
Risk Management
Risk management in a regulated eQMS is not a standalone analytical exercise — it is a live process that connects risk assessments to quality events, design decisions, supplier qualifications, and change control records. ISO 14971 is the primary risk management standard for medical device manufacturers; it requires that risk management activities be documented throughout the product lifecycle and that the risk management file be maintained current. QMSR incorporates ISO 14971 risk management principles for device manufacturers operating under 21 CFR Part 820.
An eQMS risk management module provides risk assessment templates, probability and severity scoring aligned to ISO 14971 and applicable sector frameworks, risk acceptance criteria, and mitigation action tracking. Risk records are linked to related quality events — so when a CAPA is opened or a change control is initiated that affects a risk previously assessed, the connection is visible in the record.
Design and Development
Design and development management in an eQMS governs the structured lifecycle through which new products are designed, verified, validated, and transferred to production. For medical device manufacturers, design controls are a QMSR requirement implemented through ISO 13485 Sections 7.3.2 through 7.3.10, covering design and development planning (7.3.2), inputs (7.3.3), outputs (7.3.4), review (7.3.5), verification (7.3.6), validation (7.3.7), design transfer (7.3.8), design changes (7.3.9), and the design and development file (7.3.10). The eQMS design and development module maintains this structured record set, links design outputs to risk management records, and connects design change events to the change control workflow.
Phase-gated design workflows enforce review and approval at each stage before the project advances, ensuring that design inputs are formally documented before outputs are generated, that verification evidence is complete before validation begins, and that design transfer criteria are satisfied before production release. When a design phase gate is passed and new procedures or work instructions are issued, the eQMS automatically triggers training assignments to the personnel roles affected — maintaining the connection between design progression and workforce competency documentation throughout the development lifecycle.
Supplier Quality Management
Supplier qualification and ongoing supplier quality management are regulatory requirements across all eLeaP’s primary verticals. FDA’s 21 CFR Part 211 requires pharmaceutical manufacturers to qualify suppliers of drug components, containers, and closures. QMSR requires medical device manufacturers to establish and maintain procedures for supplier controls. ISO 13485 Section 7.4 specifies purchasing controls including supplier evaluation (7.4.1), purchasing information (7.4.2), and verification of purchased product (7.4.3).
An eQMS supplier quality module maintains the approved supplier list, qualification records, supplier audit schedules and results, and corrective action records associated with supplier quality issues. When a supplier CAPA is required, the eQMS routes it through the same closed-loop workflow as internal CAPAs, maintaining consistent documentation standards regardless of whether the quality event originated internally or externally.
Training Management
Training management in an eQMS addresses the regulatory requirement that personnel performing quality-affecting work be demonstrably competent. For pharmaceutical manufacturers, 21 CFR Part 211.25 requires training records documenting the nature of training provided. QMSR requires that personnel performing quality-affecting work have the necessary training. ISO 13485 Section 6.2 requires documented competence records.
The training management capability that distinguishes an integrated eQMS from a standalone QMS is the automatic training trigger: document approvals, CAPA closures, change control implementations, and new hire onboarding all generate training assignments based on affected roles, and the quality record does not advance to the next workflow stage until training completion is confirmed. This is the training gate mechanism — the operational link between quality event management and workforce competency that eliminates the documentation gap between when a procedure changed and when the workforce was trained to it.
Regulatory Frameworks an eQMS Supports in 2026

QMSR: 21 CFR Part 820 as Amended
The Quality Management System Regulation (QMSR), effective February 2, 2026, amended 21 CFR Part 820 by replacing the legacy 1996 Quality System Regulation framework with requirements that incorporate ISO 13485:2016 by reference. The CFR Part 820 designation remains active. For medical device manufacturers operating in the United States, the QMSR is the current governing quality system regulation. An eQMS deployed in a device manufacturing environment must satisfy QMSR requirements including document control, CAPA, change control, design controls under ISO 13485 Section 7.3, and the audit trail and electronic signature requirements of 21 CFR Part 11.
ISO 13485:2016
ISO 13485 is the international QMS standard for medical device manufacturers and is incorporated by reference into QMSR. Key section mappings relevant to eQMS module design include: Section 4.2.4 (document control), Section 4.2.5 (records), Section 6.2 (personnel/competency), Section 7.3.2 through 7.3.10 (design and development), Section 7.4 (purchasing/supplier controls), Section 8.2.2 (complaint handling), Section 8.3 (nonconforming product), Section 8.5.2 (corrective action), and Section 8.5.3 (preventive action). An eQMS for a device manufacturer must be configurable to produce the records these sections require and maintain the links between them.
21 CFR Part 11
21 CFR Part 11 governs electronic records and electronic signatures in FDA-regulated environments. It does not prescribe quality system design — it establishes the technical requirements that electronic quality records must satisfy to be acceptable to FDA in lieu of paper records. Key Part 11 requirements include: audit trails that independently record the date and time of operator entries and actions that create, modify, or delete electronic records (Section 11.10(e)); electronic signatures that are unique to one individual and cannot be reused or reassigned (Section 11.100); and system controls that prevent unauthorized access and ensure record integrity. An eQMS that satisfies Part 11 natively — meaning these controls are built into the system architecture, not implemented through manual procedures — eliminates the validation burden of demonstrating Part 11 compliance through workarounds.
ISO 9001:2015 and the Transition to ISO 9001:2026
ISO 9001:2015 is the general quality management system standard applicable across industries. For manufacturers not subject to sector-specific standards (ISO 13485, IATF 16949, AS9100), ISO 9001 is the primary quality framework. An eQMS supports ISO 9001 compliance through document control (Section 7.5), competence records (Section 7.2), internal audit management (Section 9.2), nonconformity and CAPA management (Section 10.2), and management review support (Section 9.3).
ISO 9001:2026 is in development and expected to publish in 2026. The revision is anticipated to incorporate updated requirements around organizational context, risk-based thinking, and knowledge management. Quality managers should track the publication timeline and assess what configuration updates their eQMS will require when the new standard takes effect and certification transition periods begin.
GxP Requirements
GxP is the collective term for the Good Practice quality guidelines and regulations that apply across pharmaceutical, medical device, and biotech manufacturing and clinical operations. In a GxP-regulated environment, the quality system — including the eQMS — is itself subject to validation requirements. A computerized system used to create, modify, maintain, or transmit GxP records must be validated under GAMP 5 guidance to demonstrate that it does what it is designed to do and that its records are reliable. An eQMS that is pre-validated, or that provides a validation support package including IQ/OQ/PQ documentation templates, reduces the implementation timeline and validation burden for GxP-regulated organizations.
eQMS vs. Manual Quality Management: A Functional Comparison
| Function | Manual / Paper QMS | eQMS |
| Document Control | Paper files, manual versioning | Version-controlled, approval-routed, audit-trailed |
| CAPA Management | Spreadsheets, email chains | Closed-loop workflows with effectiveness verification |
| Audit Readiness | Manual assembly before each audit | Continuous record maintenance, on-demand reporting |
| Training Records | HR system or spreadsheet | Role-linked, time-stamped, retrievable by event or date |
| Change Control | Paper forms, manual routing | Automated approval routing with retraining triggers |
| Regulatory Submissions | Compiled manually from multiple sources | Structured record sets exportable for audit packages |
| Risk Management | Periodic review, static documents | Linked to quality events, continuously updated |
| 21 CFR Part 11 | Not applicable | Native electronic records and signatures compliance |
The functional differences in the table above reflect a structural difference in audit readiness. A manual QMS produces records that must be assembled, cross-referenced, and verified before an audit. An eQMS maintains those records continuously, links them to each other automatically, and produces them on demand. For a quality manager facing an unannounced FDA inspection or a certification audit with a fixed scope, the difference between those two states is the difference between a preparation period measured in weeks and one measured in hours.
Industries Using eQMS Software
Pharmaceutical and Biotech Manufacturing
Pharmaceutical manufacturers operating under 21 CFR Parts 210 and 211 and biotech organizations working under GxP requirements need an eQMS that supports batch record management, deviation handling with planned/unplanned distinction, CAPA with effectiveness verification, supplier qualification for components and contract manufacturers, and training documentation under 21 CFR Part 211.25. The eQMS must satisfy 21 CFR Part 11 for all electronic records and signatures and must be validated under GAMP 5 as a GxP computerized system.
Medical Device Manufacturing
Medical device manufacturers operating under QMSR (21 CFR Part 820 as amended) and ISO 13485 need an eQMS configured to manage design controls through the full ISO 13485 Section 7.3 lifecycle, device history records, complaint handling under Section 8.2.2, nonconforming product management under Section 8.3, and post-market surveillance linkages. The QMSR’s effective date of February 2, 2026 means that device manufacturers whose eQMS was configured to the 1996 QS Regulation framework should verify that their system configuration is current to QMSR requirements.
Life Sciences: CDMO and CRO Operations
Contract Development and Manufacturing Organizations (CDMOs) and Contract Research Organizations (CROs) operate quality systems that must satisfy the requirements of both their own regulatory obligations and those of their clients. An eQMS for a CDMO or CRO must be configurable to support multiple quality frameworks simultaneously, maintain separation between client quality records where required, and produce documentation packages that satisfy client audit requirements as well as FDA and ISO expectations.
Food and Beverage Manufacturing
Food manufacturers operating under FDA’s Food Safety Modernization Act (FSMA), HACCP principles, and GFSI-recognized certification schemes (SQF, BRC, FSSC 22000) use eQMS platforms to manage HACCP plans, supplier qualification and incoming material testing, sanitation and hygiene procedures, recall readiness documentation, and corrective action management. FSMA’s preventive controls requirements under 21 CFR Parts 117 and 507 create specific documentation obligations for hazard analysis, preventive controls, monitoring records, and corrective action records that a regulated eQMS is designed to manage.
General and Regulated Manufacturing
Manufacturers operating outside sector-specific frameworks use eQMS platforms to manage quality processes against ISO 9001:2015 and applicable OSHA requirements. For mid-market manufacturers, the eQMS addresses document control for SOPs and work instructions, nonconformance management, CAPA, supplier qualification, and the training record infrastructure required by ISO 9001 Section 7.2. The integration of quality event management with training assignment is particularly valuable in manufacturing environments where procedure changes, equipment qualifications, and corrective actions must be reflected in documented workforce competency records before the change is considered closed. Manufacturers pursuing ISO 9001 certification for the first time, or transitioning from a paper-based QMS, use the eQMS implementation as the structural foundation of their quality system buildout.
Aerospace Manufacturing
Aerospace manufacturers certified to AS9100 Revision D and suppliers operating within AS9110 or AS9120 certification scopes use eQMS platforms to manage first article inspection records, nonconformance and material review board (MRB) documentation, supplier quality and approved supplier lists, configuration management linkages, and the competency records required by AS9100 Section 7.2. For organizations tracking the IAQG’s transition to IA9100, an eQMS that can be reconfigured to accommodate revised standard requirements without full reimplementation is a practical consideration in platform selection.
Automotive Manufacturing
Automotive manufacturers and their supply chain suppliers operating within IATF 16949 certification scopes use eQMS platforms to manage the quality system requirements specific to automotive production. IATF 16949 builds on ISO 9001:2015 and adds automotive-specific requirements for core tools training (APQP, FMEA, MSA, SPC, PPAP), customer-specific requirements management, production part approval processes, and supplier quality management across multi-tier supply chains. An eQMS for an automotive manufacturer must support the competency documentation requirements for core tools personnel, nonconformance and corrective action management with 8D methodology support, and the internal audit program requirements that IATF 16949 specifies beyond the ISO 9001 baseline.
Cannabis and Hemp Manufacturing
Cannabis and hemp manufacturers operate under a complex and still-evolving regulatory environment that varies significantly by jurisdiction. In the United States, hemp-derived products regulated by FDA are subject to cGMP requirements under 21 CFR Parts 111 and 117. State-licensed cannabis operators face state-level quality and testing requirements that vary by market but increasingly reflect GMP principles. An eQMS for cannabis and hemp manufacturing must manage SOPs and batch records for cultivation, extraction, and finished product manufacturing, supplier qualification for inputs and packaging, deviation and nonconformance tracking, and CAPA workflows that satisfy both state regulatory expectations and the internal quality standards necessary for third-party testing and retail compliance programs.
The Integration Gap: Why eQMS Without LMS Creates Audit Risk
The most common documentation vulnerability in regulated manufacturing quality systems is not inadequate quality process management — it is the gap between quality records and training records. A CAPA is closed in the QMS. The corrective action involved a procedure revision. The procedure was updated and approved in the document control module. But the eQMS has no visibility into whether the affected workforce was trained to the new procedure before returning to work.
When an FDA investigator or ISO auditor asks for evidence that personnel performing a specific operation on a specific date had current training to the applicable procedure, the answer must come from a training record — not from an assertion. If the training record is in a separate system, producing that evidence requires manual cross-referencing between the QMS and the LMS. If the training records are in a spreadsheet, producing them requires reconstruction. Either scenario introduces the possibility of gaps, and gaps in training documentation are among the most consistently cited observations in FDA Form 483 reports.
An eQMS with native LMS integration eliminates this gap structurally. Document approvals generate training assignments. CAPA closures with procedure changes generate training assignments. Change control implementations generate training assignments. The training gate prevents quality workflow closure until completion is confirmed. Every connection between a quality event and a training response is preserved in the same audit trail, in the same system, without manual reconciliation.
This is not a feature comparison between eQMS platforms — it is a structural question about whether the quality system architecture is capable of producing the integrated evidence trail that regulators are increasingly asking for. Organizations evaluating eQMS platforms should ask explicitly whether training management is native to the platform or dependent on an API connection to a separate LMS, and what the validation and data integrity implications of that architectural choice are.
Selecting an eQMS: What Quality Managers Need to Evaluate
Regulatory Record Structure
The first evaluation criterion is whether the records the platform produces satisfy the specific regulatory requirements that apply to your organization. That means asking whether the audit trail captures every change to every record with a timestamp, user identity, and the before/after state of the change. Whether electronic signatures are permanently bound to the records they authenticate and meet the Part 11 uniqueness requirements. Whether document versioning preserves access to the current approved version while archiving superseded versions with their complete history. If the platform cannot demonstrate these capabilities in a validated state, it is not a regulated-industry eQMS — it is a document repository.
Validation Status and GxP Support
For organizations in GxP-regulated environments, the eQMS itself is a GxP computerized system that must be validated. Assess whether the vendor provides a validation support package including System Design Specifications, IQ/OQ/PQ protocols, and a Validation Summary Report that your quality team can use as a foundation for your own validation effort. An eQMS that arrives with no validation documentation transfers the full validation burden to the customer — a significant implementation cost that is often underestimated during platform selection.
Module Coverage and Configurability
An eQMS that covers document control but not CAPA, or CAPA but not change control, requires either manual workarounds for the uncovered processes or a second system — which recreates the integration problem the eQMS was meant to solve. Assess whether the platform covers all quality processes relevant to your regulatory obligations as native modules, not as add-on integrations that create separate validation obligations. Configurability matters for the same reason: a platform that cannot be configured to your specific workflow and approval routing requirements without custom code creates a maintenance burden every time the underlying platform is updated.
Training Integration Architecture
As discussed above, the architecture of the training integration is a quality record integrity question, not a feature preference. Ask the vendor specifically: is training management a native module within the same validated environment as the QMS, or is it an API connection to a separate platform? If it is an API connection, what is the validation status of that integration, what happens to historical training records if the integration is disrupted, and what manual reconciliation is required to produce an integrated quality-training evidence package for audit?
Implementation and Ongoing Support
eQMS implementation is a quality project, not an IT project. It involves configuring the system to your SOPs and approval workflows, migrating legacy records, validating the system in your environment, training administrators and end users, and establishing ongoing change control for system updates. Assess the vendor’s implementation methodology, their experience with organizations of comparable size and regulatory profile, and their ongoing support model — including how platform updates are handled, whether updates require revalidation, and what support resources are available during and after implementation.
Key Takeaways for Quality Managers Evaluating eQMS Platforms in 2026
An eQMS is not a software category that benefits from minimal viable feature sets. In regulated industries, the quality management system is the evidentiary infrastructure that demonstrates regulatory compliance. The platform must produce records that satisfy 21 CFR Part 11, QMSR, ISO 13485, or other applicable frameworks — not records that approximate compliance with caveats about what the system does not capture.
The practical evaluation questions for 2026 are specific. Is QMSR support current — not configured to the 1996 QS Regulation that QMSR superseded? Is the ISO 9001:2026 transition being tracked, and does the platform have a clear upgrade path? Is training management native to the platform, or does it create an integration gap? Is the validation support package sufficient for your GxP obligations, or will you need to build the validation from scratch?
Quality managers who approach eQMS selection as a compliance infrastructure decision — rather than a software procurement decision — are the ones who end up with platforms that perform under audit pressure, not just during implementation demonstrations.
Conclusion
eQMS software is the digital foundation of quality management in regulated industries — the system of record through which organizations create, approve, execute, link, and preserve the quality documentation that regulatory compliance requires. Its core modules address the specific quality process obligations of FDA, ISO, and GxP frameworks: document control that satisfies 21 CFR Part 11 and QMSR, CAPA management that closes the loop from root cause to verified effectiveness, change control that connects procedure revisions to training assignments, and audit management that maintains a continuous evidence record rather than assembling one before each inspection.
The distinction that matters most for regulated manufacturers in 2026 is whether the eQMS operates as a standalone quality record system or as an integrated platform that connects quality events to training management in a single validated environment. The former requires manual reconciliation to produce the integrated audit evidence that regulators are asking for. The latter produces it automatically — and permanently — in an audit trail that neither the organization nor the regulator needs to reconstruct.
About eLeaP
eLeaP (a product of Telania, LLC, founded 2002) provides an integrated Quality Management System and Learning Management System built for regulated industries. The platform’s native QMS+LMS architecture is the operational basis of its eQMS design: CAPA events, document revisions, and change control actions automatically trigger training assignments, and quality records do not close until training completion is confirmed through the training gate mechanism.
eLeaP serves mid-market organizations in pharmaceutical manufacturing, medical device, biotechnology, general manufacturing, food and beverage, aerospace, and cannabis — with compliance infrastructure supporting QMSR (21 CFR Part 820), ISO 13485, 21 CFR Part 11, ISO 9001, and GxP requirements. Learn more at quality.eleapsoftware.com.