QMS for CDMO and CRO Organizations
The eQMS that unifies multi-client GxP quality processes and personnel training in one validated system.


GxP Quality Management for Contract Operations
eLeaP is the only Quality Management System with enterprise Learning Management built in — purpose-built for CDMOs and CROs operating under FDA, ICH Q10, and EU GMP requirements. Where other platforms require a separate LMS integrated via API, eLeaP automates personnel qualification across every quality process from a single validated system. → Get a Demo.
How It Works:
When GMP procedures are revised and approved, they automatically become training assignments with completion tracking. Tech transfer milestones deploy required training to affected roles. Risk assessments trigger mitigation training when control measures are updated. CAPAs and deviation investigations drive targeted corrective training with documented effectiveness verification — all fully traceable for FDA inspections and client audits. Our pre-validated platform deploys in weeks, with built-in 21 CFR Part 11 compliance and templates aligned to FDA, ICH, EU GMP, and ISO requirements.
Controlled Document Management for GMP Contract Operations
CDMOs manage controlled documents across multiple client product lines, each with distinct procedure sets, approval authorities, and revision histories. A single document control failure — a superseded SOP on the manufacturing floor, an unapproved change to a batch record template — can trigger a client audit finding, a deviation report, or an FDA observation. eLeaP prevents this by making document control and training deployment a single automated event.
Document Control Built for Multi-Client GMP Operations
Manage SOPs, batch manufacturing records, master batch records, work instructions, client-specific specifications, and quality policies with version-controlled review and approval workflows. Electronic signatures meet 21 CFR Part 11 requirements. Complete audit trails support FDA inspection readiness and client audit readiness at all times.
Access controls enforce which personnel can view or edit documents associated with specific clients or product lines. When a procedure goes effective, training automatically deploys to every affected role — no manual assignment, no spreadsheet tracking. Read-and-understood acknowledgments, comprehension assessments, and training matrices maintain themselves.
When a client auditor reviews your documentation records, everything is in one place, segregated by client scope, and traceable from document approval through training completion. Learn more. Get a Demo.
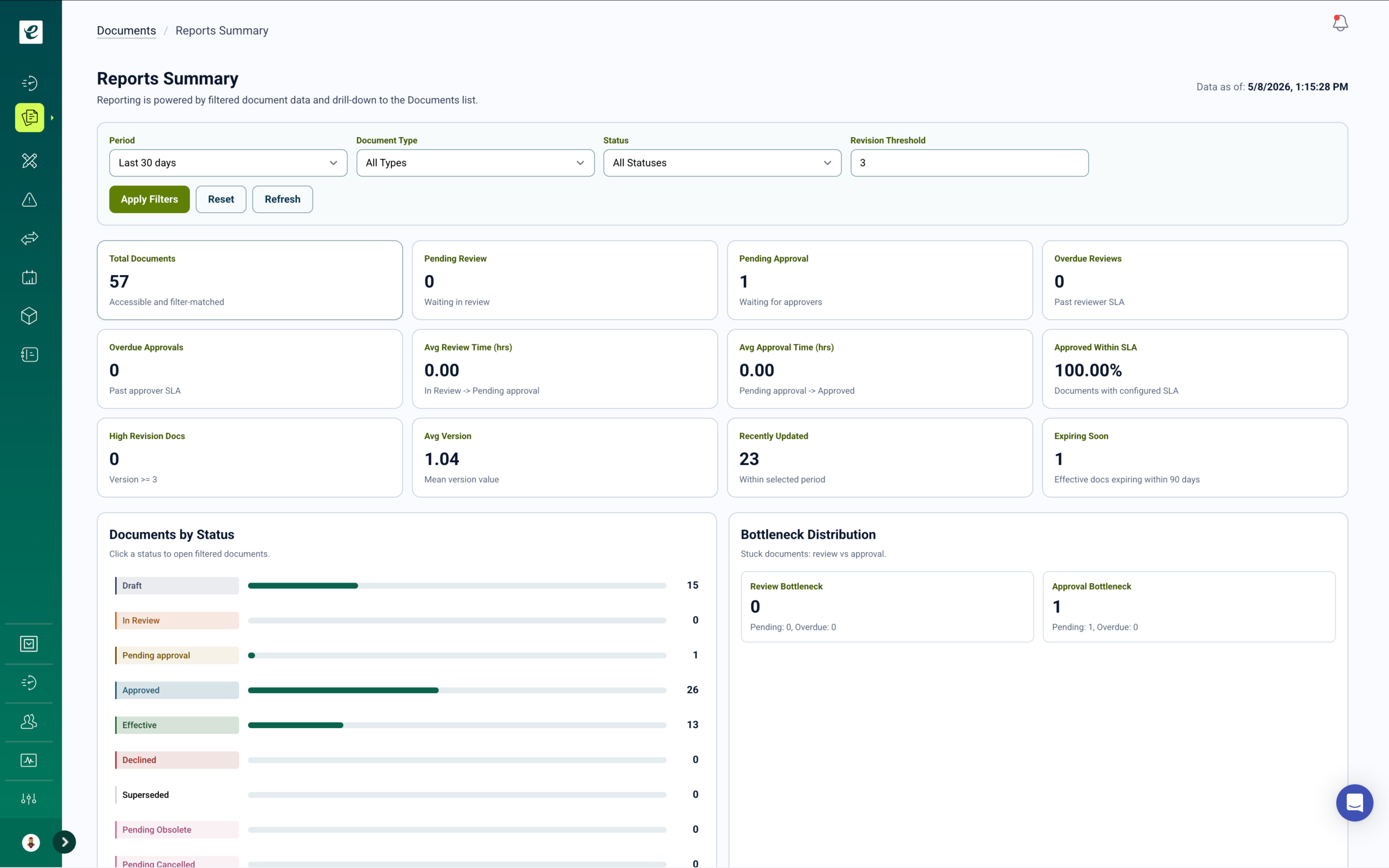
Tech Transfer and Process Development Quality Management
Tech transfer is one of the highest-risk activities a CDMO undertakes. Procedures written by the innovator must be translated into the CDMO’s operational context, validated in the receiving facility, and demonstrated to reproduce the originator’s process performance. Each phase gate generates new controlled documents, updated risk assessments, and training obligations for the personnel who will execute the commercial process. Without a system that connects these events, the quality record is incomplete before the first commercial batch runs.
Phase-Gated Tech Transfer with Integrated Training
eLeaP’s development module manages tech transfer and process development through structured phase gates aligned to ICH Q10 and client-specified transfer protocols. Each phase gate requires completion of defined deliverables — process characterization reports, equipment qualification protocols, analytical method transfers, validation protocols — before the project advances.
When a phase gate is passed and new procedures or work instructions are issued, training automatically deploys to the production, quality, and analytical roles that will execute the validated process. Personnel cannot advance through quality workflows dependent on the transferred process until training completion is confirmed. The tech transfer record — from initial feasibility through process validation and commercial release — is maintained in a single traceable system.
Process development reports, scale-up documentation, and validation summary reports are stored in the same controlled environment as the SOPs they support, with complete version history and approval records inspection-ready at all times.
Get a Demo.
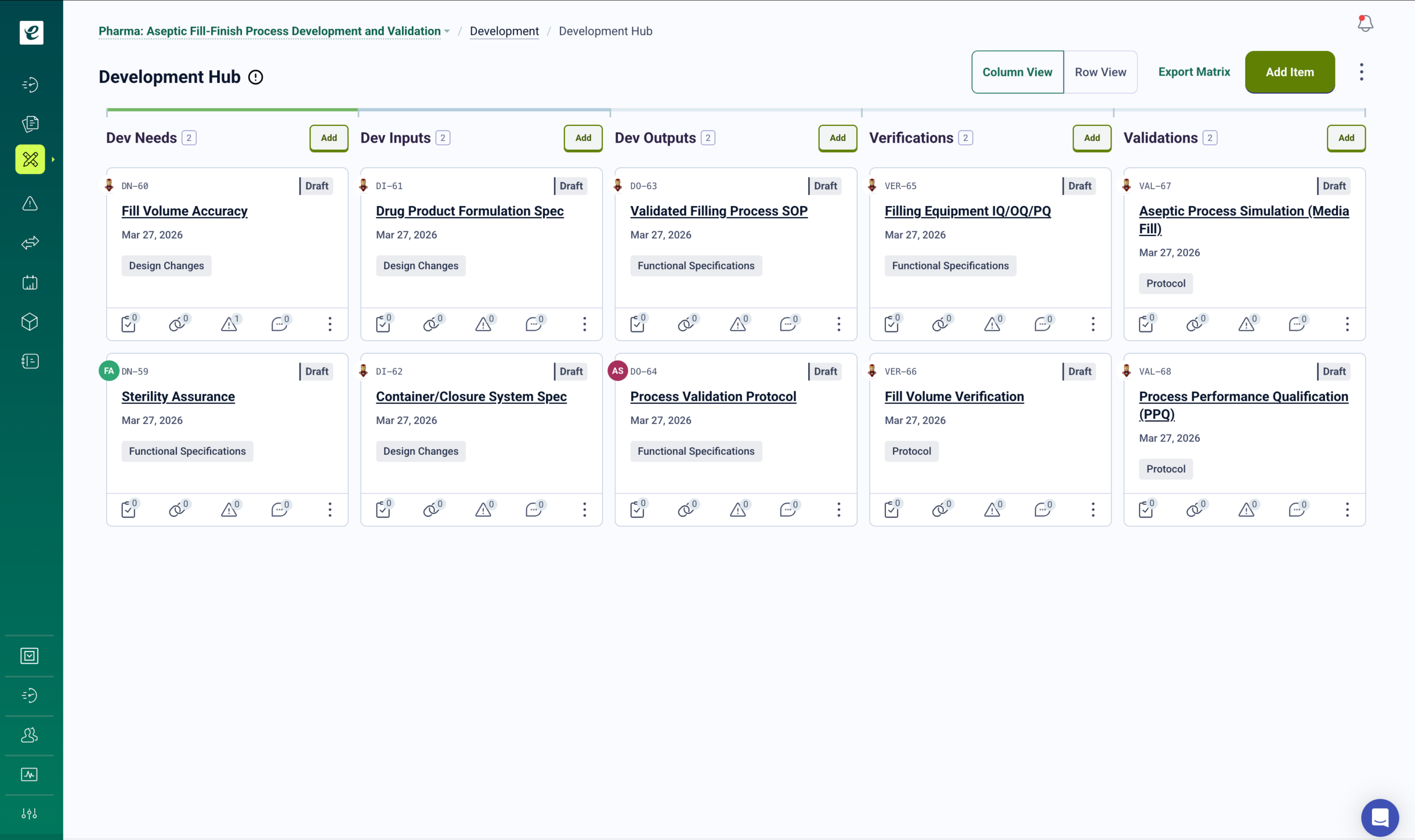
ICH Q9 and EU GMP Risk Management for Contract Operations
ICH Q9 quality risk management applies across all stages of the pharmaceutical lifecycle, including contract manufacturing. EU GMP Annex 1 (2022 revision) significantly expanded risk management requirements for sterile manufacturing CDMOs, requiring a formal Contamination Control Strategy documented as part of the pharmaceutical quality system.
eLeaP’s risk management module supports both frameworks from a single validated environment.
Risk-Based Quality Management Across Client Product Lines
Conduct FMEAs for manufacturing process steps, assess risk for equipment changes and tech transfer activities, and document control measures with probability and severity scoring aligned to ICH Q9 methodology. Risk assessments are scoped by client or product line, maintaining separation between client risk records in the same platform.
When risk assessments identify new control measures or procedural safeguards, training automatically deploys to affected personnel. Root cause analysis connects quality events to training history — identifying whether issues stem from knowledge gaps, procedural failures, or process deficiencies. Risk reviews, control effectiveness monitoring, and CAPA linkage are maintained in a single traceable system. For sterile manufacturing CDMOs, the Contamination Control Strategy is managed as a living document within the risk module, linked to the cleaning validation records, environmental monitoring data, and personnel hygiene training records that support it. Get a Demo.

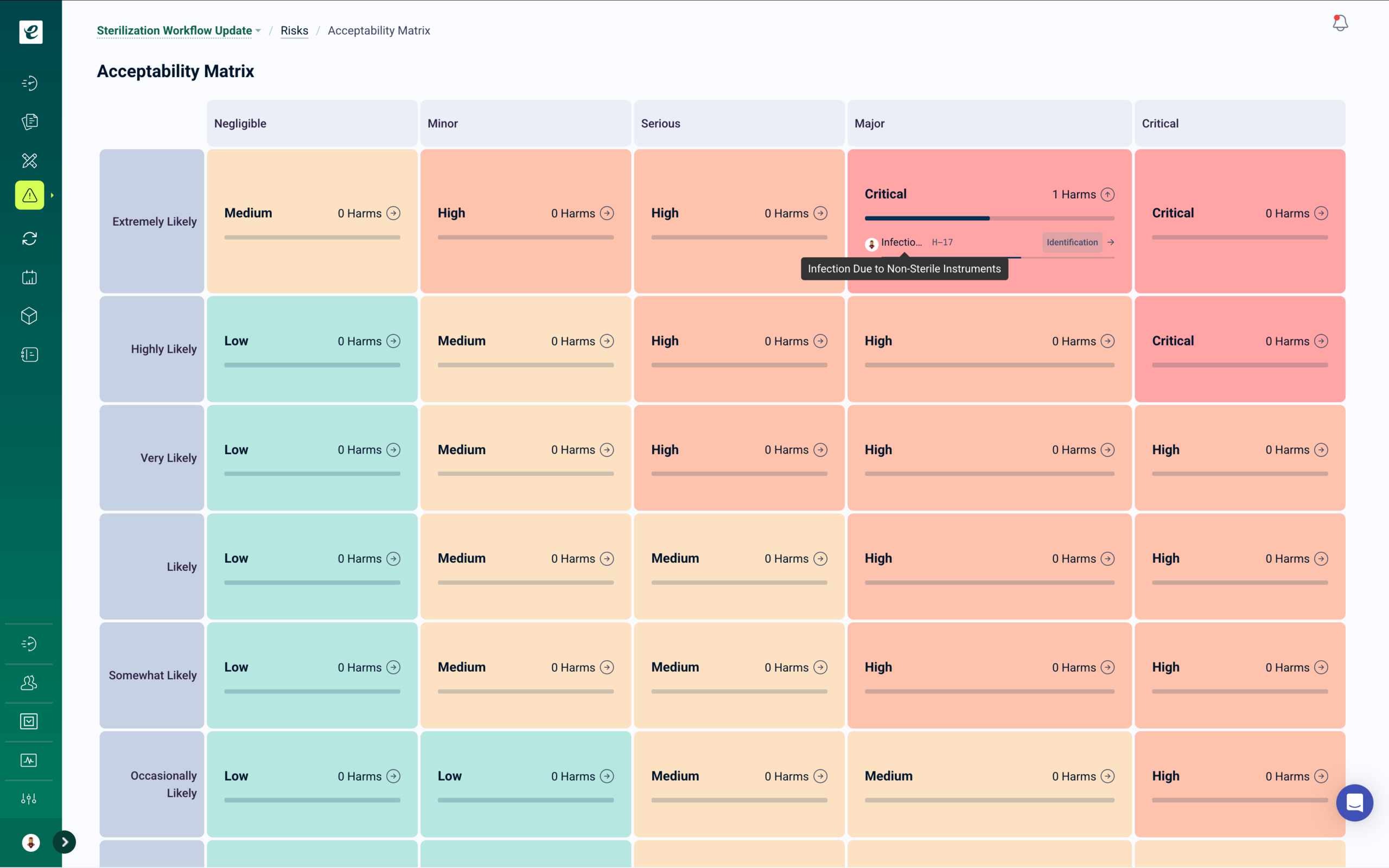
GxP Change Control with Automatic Training
Manufacturing process changes, procedure revisions, equipment modifications, and tech transfer scope changes automatically trigger targeted training for affected personnel — with client notification workflows and documented implementation verification before changes go live.
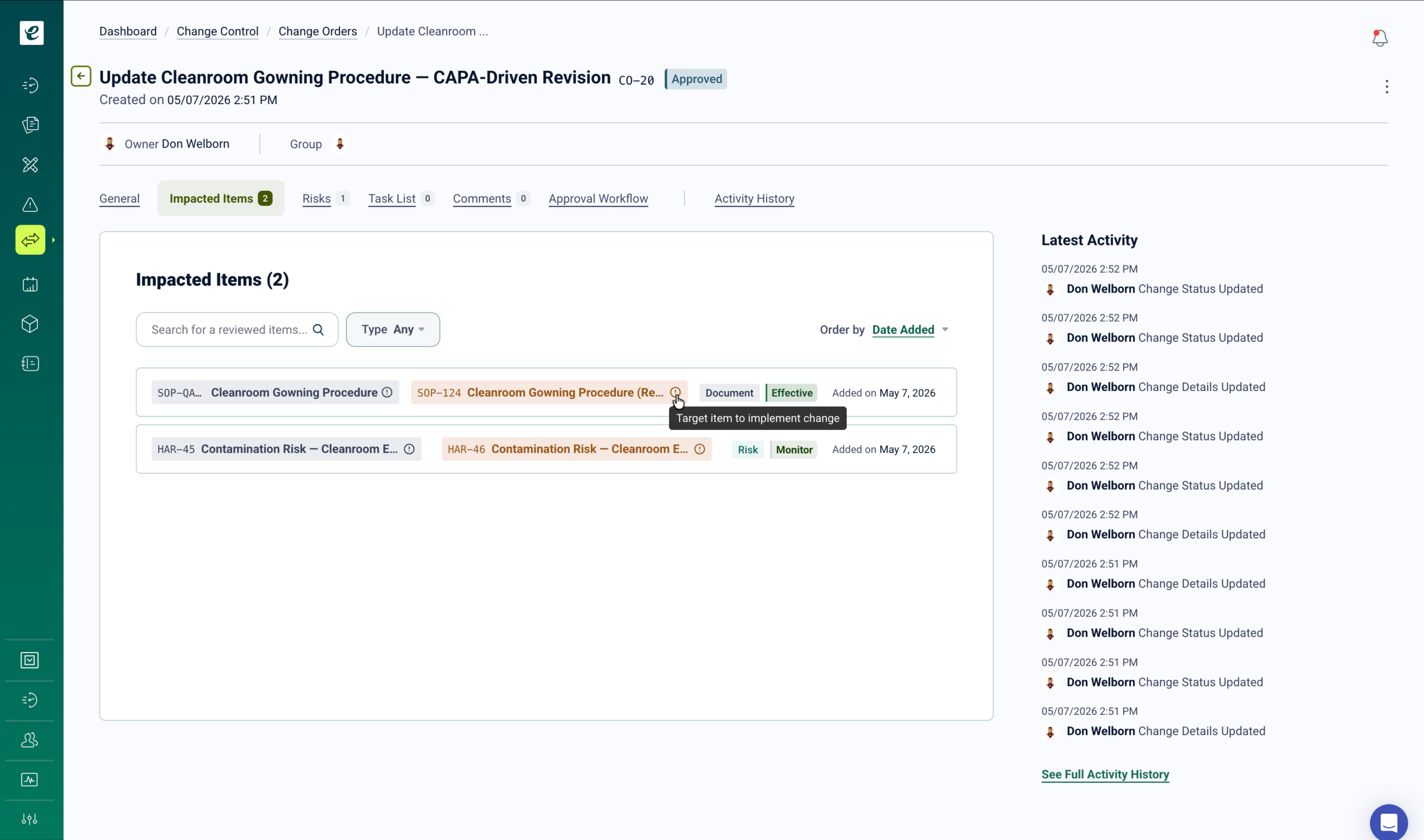
In GxP contract manufacturing, every change requires documented impact assessment, appropriate approval, and verified implementation — including personnel training before the change is activated. Changes initiated by the client must be reviewed for CDMO manufacturing impact. Changes initiated by the CDMO must be communicated to clients per quality agreement terms. eLeaP’s change control module manages both directions of this obligation from a single controlled workflow.
Change Control That Closes the Training Gap
When a change is approved, training deploys immediately to all roles identified in the impact assessment. Smart assignment rules match personnel to required training based on their functions and the specific product lines affected. Competency assessments verify understanding. Change implementation is confirmed only after training completion is documented.
For changes that require regulatory reporting — post-approval changes notified to the client’s regulatory authority under ICH Q12 pathways, or manufacturing changes that trigger site change supplements — the change control record provides the documentation package supporting the submission. Complete change history, impact assessments, approval records, and training verification are maintained in a single traceable record.
Client-specific change notification workflows are configurable within eLeaP, ensuring that quality agreement obligations around change communication are fulfilled as part of the change control process, not as a separate manual step.
Get a Demo.
CAPA, Deviation, and OOS Management Across Client Operations
21 CFR 211.192 requires investigation of any unexplained discrepancy or failure to meet specifications. For CDMOs, deviations and OOS results carry dual significance: they generate internal quality obligations and they trigger client notification requirements defined in the quality agreement. eLeaP’s events management module handles deviations, CAPAs, OOS investigations, and audit findings — and automatically connects each to training when investigation identifies a personnel competency gap.
Close the Loop Between Quality Events and Personnel Qualification
When a deviation identifies a procedural misunderstanding or an OOS investigation points to analyst error, training assignments deploy automatically. CAPA closure requires documented training completion and effectiveness verification — the record does not close until both are confirmed. Every event record is scoped to the applicable client product line, with access controls that maintain client data separation.
Audit findings from internal audits, client audits, and regulatory authority inspections are captured in the same events workflow, with structured finding classification, CAPA linkage, and follow-up audit scheduling. When a client auditor reviews your corrective action records, the complete chain from finding to root cause to corrective action to training verification is visible in a single record.
ICH Q10 Section 3.2.2 CAPA requirements are satisfied through the closed-loop workflow: problem identification, root cause analysis, corrective action with assigned owners and due dates, implementation verification, and effectiveness check at a defined post-closure interval. Get a Demo.
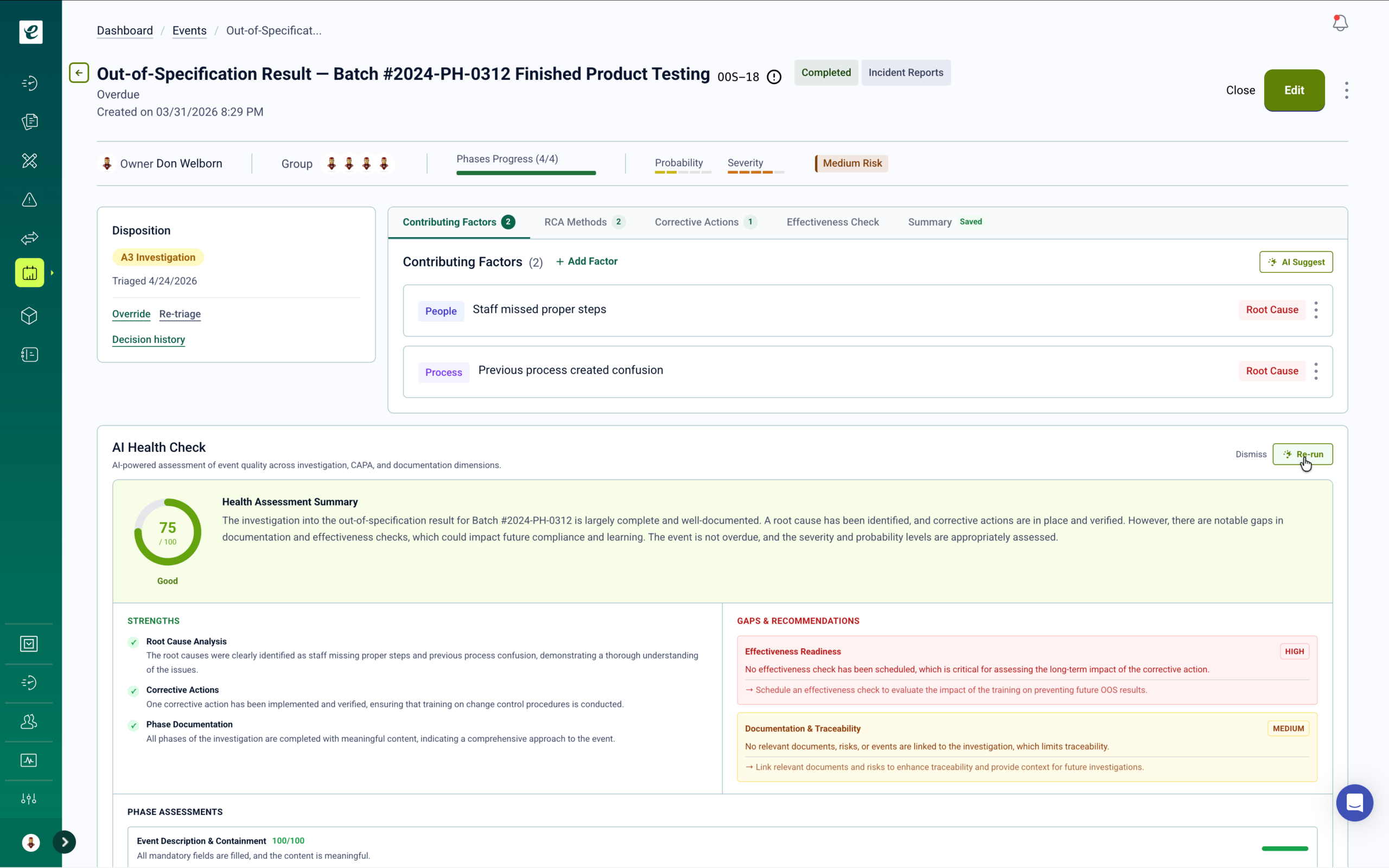
Supplier Quality Management for GMP Contract Supply Chains
Risk-based supplier qualification, quality agreement management, and audit tracking — with automatic training triggers when supplier audits identify gaps in internal receiving, testing, or qualification procedures.
CDMOs procuring APIs, excipients, packaging components, and laboratory consumables on behalf of client products must maintain supplier qualification records that satisfy both the CDMO’s own GMP obligations and the requirements specified in client quality agreements. Under ICH Q10 and 21 CFR Part 211, the contract giver retains ultimate responsibility for material quality — but the CDMO executes and documents the supplier oversight activities. eLeaP’s supplier management module maintains the complete qualification record in the same validated environment as all other quality processes.
GMP Supply Chain Quality Management for Contract Operations
Maintain the approved supplier list, qualification records, audit schedules and results, quality agreement documentation, and incoming material inspection records within eLeaP’s supplier module. Supplier performance metrics, qualification status, and audit findings are tracked in a system linked directly to the broader quality record — not in a separate supplier portal that creates a documentation gap.
When supplier audits identify gaps in internal receiving, testing, or qualification procedures, training automatically deploys to affected internal teams. Supplier performance trends visible across the quality system connect supply chain inputs to deviation rates and OOS frequencies, enabling risk-based supplier management decisions grounded in quality data rather than audit schedules alone.
For clients with specific supplier qualification requirements — approved supplier lists, incoming testing specifications, certificate of analysis review criteria — those requirements are configurable within eLeaP’s supplier module by client or product line, satisfying quality agreement obligations within the same workflow. Get a Demo.

GMP and GCP Training Compliance — Built In. Not Added On.
Enterprise learning management is part of the platform — not integrated via API, not managed in a separate system. Quality events trigger training automatically. Personnel qualification is verified in real time.
21 CFR Part 211.25 requires that every person engaged in pharmaceutical manufacturing have documented education, training, and experience sufficient to perform their assigned functions. For a CDMO managing multiple client product lines with changing procedures, recurring quality events, and ongoing tech transfer activities, meeting this requirement across a shared workforce requires more than a training system that talks to your QMS. It requires a system where quality and training are the same system.
Qualification matrices show who is trained on what, in real time, across every product line, procedure, and piece of equipment — by client scope. When a regulator or client auditor asks for training records for the analysts who ran a specific batch, the answer is immediate. No spreadsheets. No reconciliation between systems. No manual compilation before the auditor arrives.
When a client quality agreement requires demonstration that CDMO personnel are qualified for the specific procedures governing their product, eLeaP produces that evidence automatically — time-stamped, role-linked, and scoped to the client’s product line.
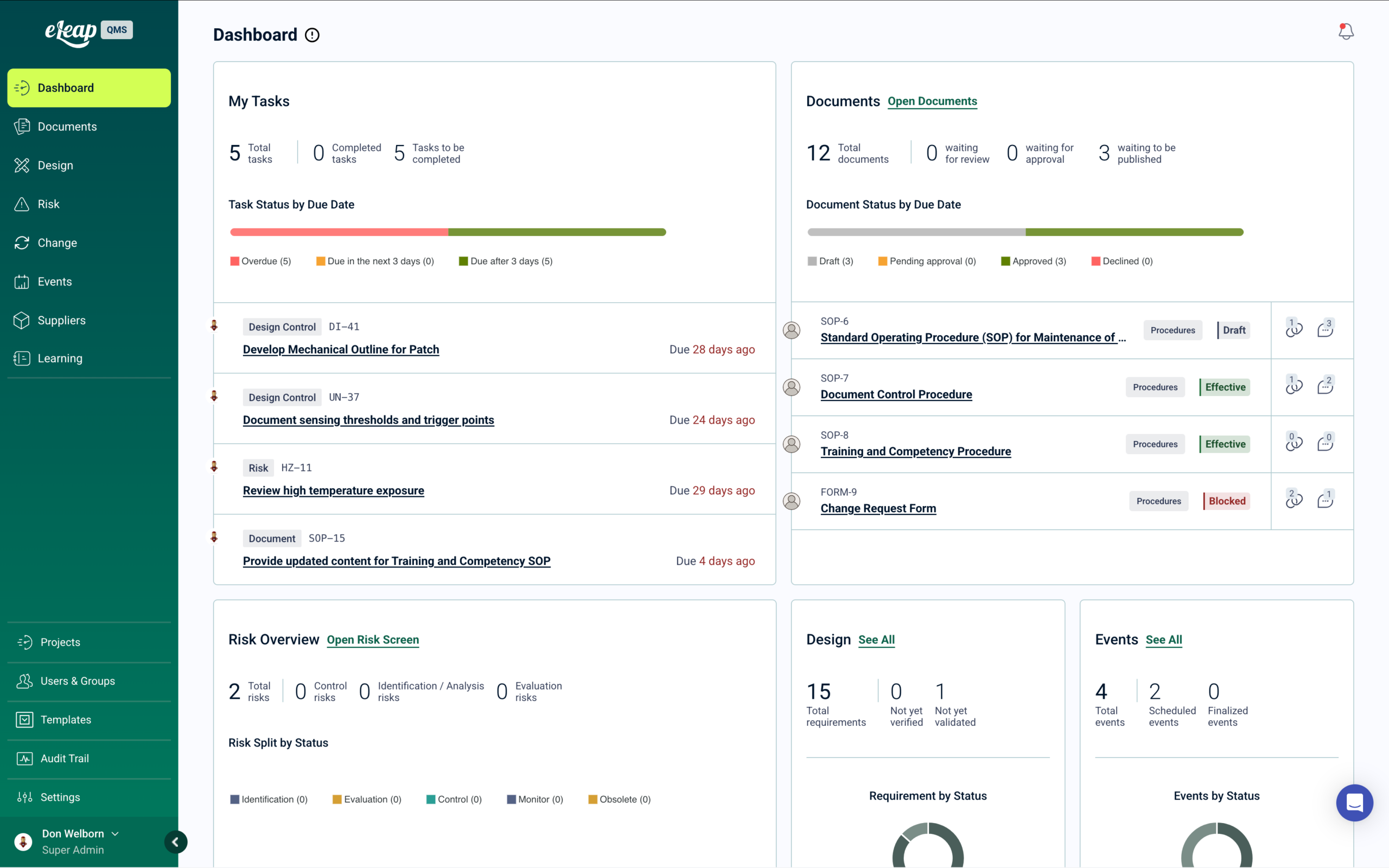
Building GxP Compliance Into Every Workflow
FDA inspections and client audits of CDMOs consistently cite training deficiencies, inadequate documentation of personnel qualifications, and gaps between quality events and corrective training. eLeaP eliminates these gaps by making training an automatic output of every quality process — not a manual step someone has to remember.
-
Automatic Training Triggers
Procedure approvals, tech transfer milestones, deviation closures, and CAPA actions automatically deploy training to the right personnel at the right time.
-
Real-Time Qualification Verification
Personnel qualification status is visible across all client product lines, procedures, and equipment — no spreadsheets, no manual tracking.
-
Inspection-Ready by Design
Complete audit trails connect quality decisions to training records. When inspectors or client auditors ask, you answer immediately.
Quality and Training Working as One System
GxP contract operations should give quality leaders the tools to eliminate compliance blind spots while giving manufacturing and analytical personnel timely, relevant training connected directly to the procedures governing their work. eLeaP gives CDMOs and CROs both — in one validated system. Get a Demo.
-
Continuous Training Integration
Every quality process — documents, development, risk, change, events, suppliers — automatically drives training when it matters.
-
Connected Quality Data
All modules work as one system. No disconnected client records. No manual handoffs. Complete audit trails across every product line.
-
Competency-Driven. Root Cause Analysis
Investigations connect training history, competency assessments, and quality outcomes — identifying whether issues stem from knowledge gaps or process failures.
-
Here, When You Need Us
Our quality experts support implementation, process optimization, and compliance program development for CDMO and CRO organizations.
Frequently Asked Questions: QMS for CDMO and CRO Operations
What regulatory frameworks does a CDMO QMS need to support?
A CDMO QMS must support the frameworks applicable to its clients’ products. For pharmaceutical CDMOs, this includes FDA 21 CFR Parts 210 and 211, ICH Q10, EU GMP, and 21 CFR Part 11 for electronic records. For device CDMOs, QMSR (21 CFR Part 820 as amended effective February 2, 2026) and ISO 13485 apply. Many CDMOs operate under multiple frameworks simultaneously, requiring a QMS configurable by client or product line without creating separate quality system silos.
How does a CDMO QMS manage quality records for multiple clients?
A purpose-built CDMO QMS provides record segmentation by client or product line, with access controls that enforce which personnel can view or edit records associated with specific clients. Audit packages can be assembled by client scope, allowing the CDMO to respond to client audits without exposing other clients’ data. eLeaP’s architecture supports this segmentation natively within a single validated platform.
What is the difference between a CDMO and a CRO from a QMS perspective?
CDMOs perform manufacturing operations subject to GMP regulations, generating batch records, deviation reports, and release documentation. CROs conduct clinical trials, analytical testing, or research under GCP or GLP frameworks. Both require documented quality systems, but the applicable regulations and record types differ. eLeaP is configurable to support GMP, GCP, and GLP record structures and the different training requirements each framework imposes.
How does tech transfer fit into a CDMO quality management system?
Tech transfer is a structured quality process in which manufacturing processes developed by an innovator are transferred to the CDMO’s facility, validated, and demonstrated to reproduce the originator’s process performance. Each tech transfer phase gate generates new controlled documents, updated risk assessments, and training obligations. eLeaP’s development module manages these phase gates, links documents and risk records to training assignments, and maintains the complete transfer record from feasibility through commercial release.
How does 21 CFR Part 11 apply to a CDMO’s quality records?
Any electronic record created, modified, maintained, archived, retrieved, or transmitted under FDA requirements must satisfy 21 CFR Part 11. For a pharmaceutical CDMO, this includes batch records, deviation records, CAPA records, and training records. Part 11 requires audit trails capturing the date, time, and identity of each person creating or modifying records, and electronic signatures permanently bound to the records they authenticate. eLeaP satisfies Part 11 requirements natively — these controls are built into the platform architecture.
What training documentation does a CDMO need to maintain for GMP compliance?
Under 21 CFR Part 211.25, CDMO personnel performing GMP operations must be documented as qualified for their assigned tasks. Training records must document what training was provided, when it was completed, and to what procedure version. When SOPs are revised, records must show that affected personnel were trained to the new version before performing the work. eLeaP automates these assignments and maintains the complete competency record — by client product line, by procedure version, by personnel role — without manual tracking.
How does ICH Q10 Section 2.7 apply to CDMOs?
ICH Q10 Section 2.7 addresses outsourcing arrangements, requiring that both the contract giver and the CDMO have written agreements covering quality responsibilities, that the CDMO’s quality system satisfies the required GMP standard, and that the contract giver maintains oversight of CDMO quality performance. The CDMO’s eQMS must support the CAPA, change management, and knowledge management elements of ICH Q10 and produce the documentation that demonstrates these obligations are being fulfilled to client auditors.
Can a single eQMS platform serve both CDMO manufacturing and CRO clinical operations?
Yes, provided the platform is configurable to support different record structures, regulatory frameworks, and access control models for each business unit. The key criterion is whether the QMS maintains genuine record separation between manufacturing and clinical operations while enabling consolidated quality oversight. eLeaP’s configurable module structure and role-based access controls support both operational contexts within a single validated platform.
About eLeaP
eLeaP (a product of Telania, LLC, founded 2002) provides an integrated Quality Management System and Learning Management System built for regulated industries. For CDMOs and CROs, eLeaP’s native QMS+LMS architecture means that GMP procedure revisions, CAPA corrective actions, tech transfer milestones, and deviation investigations automatically trigger training assignments — and quality records do not close until training completion is confirmed through the training gate mechanism.
eLeaP supports multi-client record management, 21 CFR Part 11-compliant electronic records and signatures, ICH Q10 CAPA and change management, EU GMP Annex 11-compliant system controls, and GCP/GLP quality processes — in a single validated platform. Learn more at quality.eleapsoftware.com.